Ancient DNA assembly
Part 2 of the Museomics Workshop
This tutorial is Part 2 of the Museomics Workshop (CVZoo XIV 2025, University of São Paulo, Brazil). All software used in this tutorial were previously installed during Part 1 of the Museomics Workshop: check it here!
Before starting the tutorial, download the resources here. If you completed Part 1, the resources were previously downloaded (no need to download them again).
What is museomics?
Museomics is the application of genomic and bioinformatic methods to museum specimens, such as skins, bones, teeth, feathers, shells, pinned insects, and herbarium samples. These specimens are typically from extinct or historically collected organisms and were not preserved with DNA analysis in mind. Paleogenomics is the study of very old biological material preserved under natural conditions, such as subfossils from caves (e.g., Neanderthals and Denisovans) and permafrost-preserved tissues (e.g., Ice Age megafauna). Archival DNA refers to DNA recovered from any archived biological material, including DNA from museum specimens (historical DNA; hDNA), typically ranging from ~50 to 200 years old, as well as much older biological material (ancient DNA; aDNA), which can range from thousands to millions of years old. However, in the literature, these terms are sometimes used interchangeably. As a shorthand, we will use only the terms “museomics” and “hDNA” throughout this tutorial.
Challenges and solutions
Working with hDNA presents several well-known challenges. DNA molecules are typically highly fragmented and present in very low quantities, with low endogenous content due to the predominance of environmental and microbial DNA. In addition, post-mortem damage—especially cytosine deamination leading to C→T and G→A substitutions—introduces characteristic errors, and contamination from modern DNA (human, microbial, or laboratory sources) can overwhelm authentic sequences.
The aforementioned challenges have driven the development of both wet-lab and computational solutions. In the laboratory, dedicated hDNA facilities with strict contamination control, optimized extraction protocols, and the use of second-generation sequencing (NGS) enable the recovery of short DNA fragments (reads). Enzymatic treatments such as USER (UDG + Endonuclease VIII) can partially or fully remove deaminated cytosines and abasic sites, reducing post-mortem mutation. Single-stranded libraries can minimize the loss of endogenous content. In silico, bioinformatic approaches explicitly model ancient DNA properties, including the use of damage profiles to identify and authenticate ancient molecules, in silico contaminant deletion, iterative mapping strategies to improve alignment of short and divergent reads, and filtering steps to minimize contamination. Overall, these strategies have provided advances in museomics.
Below, you will learn how to preprocess, assemble, and post-process hDNA reads.
Preprocessing
There are three FastQ files in museomics/part2/1_raw_reads, all sequenced from Dendropsophus rhea MZUSP 14458. Initially, we merge these single-end FastQ files that represent the same sample. Assuming you are in museomics/part2/1_raw_reads, run:
# Create a new directory
mkdir ../2_merged; cd ../2_merged
# Merge reads from multiple lanes but same sample
cat ../1_raw_reads/Drhea* > Drhea_merged.fastq
# Rename headers to avoid redundancy
conda activate seqkit
seqkit rename Drhea_merged.fastq > Drhea_renamed.fastq
conda deactivate
Quality assessment
FastQC is an essential quality control (QC) tool used in bioinformatics to assess the raw data generated by high-throughput sequencers (like Illumina). Developed by Simon Andrews at Babraham Bioinformatics, it provides a quick, visual overview of whether your sequencing data has potential problems, biases, or contamination before you proceed to downstream analysis.
Assuming you are in museomics/part2/2_merged, run:
conda activate fastqc
fastqc *renamed.fastq
conda deactivate
The output HTML file Drhea_renamed_fastqc.html presents plots that can be visualized in the browser.
Exercise 1 Check the file
Drhea_renamed_fastqc.html. Describe and create hypotheses to explain the distributions of (1) per base sequence content, (2) sequence length, and (3) sequence duplication levels.Show answer
(1) The distribution of per base sequence content presents a high proportion of G relative to A, C and T at the ends of reads. A explanation is the high level of PolyG at the ends of reads. PolyG commonly indicate signal decay during the end of sequencing (and not a real G) because G is represented by no color in some platforms.
(2) The sequence length distribution reveals a bicaudal distribution. The first peak around 20-30 bp likely represents fragmented hDNA reads, whereas the the second peak around 98-100 bp likely represents modern DNA contaminants.
(3) The sequence duplication levels reveals that some sequences are duplicated. Possible explanations include PCR duplicates not removed and fragments from different regions of the genome randomly sharing the same sequence.
Trimming
Illumina libraries present adapters that must be trimmed before downstream analyses. Furthermore, hDNA fragments are often very short (sometimes <50 bp). As such, finding unusually long reads (e.g., >200–300 bp in a highly degraded sample) is actually a major red flag. Conversely, while museomics depends on short fragments, there is a limit to how short a read can be while still being useful. Very short fragments (< 20 bp) can introduces massive noise into your data, making it impossible to accurately map reads (assemble hDNA) in next steps.
Assuming you are in museomics/part2/2_merged, run:
# Create a new directory
mkdir ../3_cutadapt; cd ../3_cutadapt
# Trim adapters and reads out of size range
conda activate cutadapt
cutadapt -O 4 -a AGATCGGAAGAGCACACGTC -m 21 -M 90 -o Drhea_cutadapt.fastq ../2_merged/Drhea_renamed.fastq
# Assess quality of reads
conda activate fastqc
fastqc *fastq
conda deactivate
Exercise 2 Check the files
Drhea_cutadapt_fastqc.htmlandDtritaeniatus_cutadapt_fastqc.html. Compare both reports with those before trimming. Describe what happened with the per base sequence quality, sequence distribution, overrepresented sequences, and adapter content.Show answer
The per base sequence quality improved, the sequence distribution is right-skewed with one peak between 20-30 bp, and adapters and other artifacts like PolyG were deleted.
Deduplicating
Deleting PCR duplicates is necessary because hDNA libraries originate from a very small number of unique, degraded template molecules that require extensive PCR amplification to reach detectable levels. Consequently, many PCR duplicates are present, which, if not removed, falsely inflate sequencing coverage. Leaving these duplicates in your analysis can lead to significant errors such as false-positive SNP calls, compromising the accuracy of population genetic and phylogenetic inferences.
Assuming you are in museomics/part2/3_cutadapt, run:
# Create a new directory
mkdir ../4_tally; cd ../4_tally
# Delete PCR duplicates
conda activate tally
tally -i ../3_cutadapt/Drhea_cutadapt.fastq -o Drhea_tally.fastq --nozip --with-quality
# Assess quality of reads
conda activate fastqc
fastqc *fastq
conda deactivate
Exercise 3 Check the files
Drhea_tally_fastqc.htmlandDtritaeniatus_tally_fastqc.html. Compare both reports with those before deduplicating. Describe what happened with the total number of sequences (available in Basic Statistics) and the sequence duplication levels.Show answer
The total number of sequences slighly reduced because duplicates were deleted.
Decontaminating
In museomics, deleting contaminants using FastQ Screen is a critical step for validating the authenticity of historical DNA and preventing the analysis of modern biological noise. Because DNA from museum specimens is often highly degraded, non-target DNA—originating from the museum environment, laboratory handling (human DNA), or even the reagents themselves—can easily outnumber the endogenous DNA of interest. FastQ Screen allows researchers to map their sequencing reads against a panel of diverse reference genomes (e.g., human, common bacteria, or potential contaminants like PhiX) to quantify and then filter out sequences that do not belong to the target species.
Assuming you are in museomics/part2/4_tally, run:
# Create a new directory
mkdir ../5_fastqscreen; cd ../5_fastqscreen
FastqScreen is a script to map reads against a database of potential contaminant genomes and delete the mapped reads. The first step is creating the database of contaminants. It is located in museomics/part2/FastqScreenGenomes.zip. Unzip this directory and check the subdirectories containing indexed contaminant genomes. The file fastq_screen.conf should be edited in each analysis to specify the path for Bowtie2 aligner (L12; check it using the command which bowtie2), number of threads (cores) for parallel analyses (line 21), and contaminant databases (E. coli, vector, adapters, and Paraburkholderia).
In this tutorial, indexing genomes or editing the configuration file are not necessary for FastqScreen analyses (unless the paths in the configuration file are not working). Moreover, human genome was ignored as a potential contaminant to accelerate analyses.
First, in library preparation (wet lab), indexing is the process of adding a unique "ID tag" to your DNA fragments in multiplex libraries (i.e. containing multiple samples to be sequenced together). Without these tags, you would have millions of sequences but no way to know which read came from which specimen. After sequencing, the sample of each read is identified during a computational step called demultiplexing.
Second, in read mapping (bioinformatics), you need to "index" your reference genome. This is very similar to the index at the back of a textbook. One possibility is creating a separate set of files (e.g. hash table) that split the reference genome into k-mers and inform their correspondent positions in the genome. Another tool for indexing is the Burrows-Wheeler Transform (BWT), in which it reorders the genome millions of times (so that similar characters are grouped together) and keep the last column. If you tried to align a read by searching the 3-billion-base human genome from start to finish for every single read, it would take years. With an index, the software can "jump" directly to the potential matching locations in milliseconds.
conda activate fastqscreen
# Define the configuration file (don't copy and paste the command below, check the path in your computer)
config="/mnt/c/Users/Aluno/Downloads/CVZoo-20260120T180729Z-3-001/CVZoo/FastqScreenGenomes/fastq_screen.conf"
# New contaminants might be indexed with the following command: bowtie2-build contaminant_name.fasta name_index
fastq_screen --nohits --aligner bowtie2 --conf $config ../4_tally/Drhea_tally.fastq
# If it fails, edit the path of each database and the commands above again
nano $config
# Assess quality of reads
conda activate fastqc
fastqc *fastq
conda deactivate
Exercise 4 Open the files
Drhea_tally_screen.htmlandDtritaeniatus_tally_screen.htmlin your browser. Which contaminant presents more hits?Show answer
Paraburkholderia.
Assembly
Sanger sequencing generates few contigs (up to 900 bp) based on chromatogram peaks. In contrast, next-generation sequencing (NGS) handle millions of short reads (usually 50–150 bp) with low error rates (in second-generation) or a few long reads (10k-1M bp) with high error rates (in third-generation). NGS read mapping (alignment) acts like a “shortcut” by using an existing reference genome as a blueprint to organize these tiny fragments. NGS de novo assembly is the most complex approach, building a entirely new genome from scratch by finding overlaps between reads without any external guide—a task that is often impossible for hDNA due to the lack of sufficient overlap between highly fragmented, short reads.
As such, in museomics, we use second-generation sequencing and short read mapping.
Indexing and linear mapping
You will run BWA using two samples (D. rhea and D. tritaeniatus) and three references (the mitochondrial 12S and the nuclear 28S and RAG1 of D. microcephalus).
Assuming you are in the directory museomics/part2/5_fastqscreen, let’s index the reference sequences.
# Create a new directory
mkdir ../6_bwa; cd ../6_bwa
conda activate bwa
mkdir bwa_index; cd bwa_index
# Index reference of 28S
mkdir Dmicrocephalus_28s; cd Dmicrocephalus_28s
bwa index -p Dmicrocephalus_28s ../../../reference/Dmicrocephalus_28s.fas
# Index reference of 12S
mkdir ../Dmicrocephalus_12s; cd ../Dmicrocephalus_12s
bwa index -p Dmicrocephalus_12s ../../../reference/Dmicrocephalus_12s.fas
# Index reference of RAG1
mkdir ../Dmicrocephalus_rag1; cd ../Dmicrocephalus_rag1
bwa index -p Dmicrocephalus_rag1 ../../../reference/Dmicrocephalus_rag1.fas
cd ../..
Now, we can map reads to the indexed references. We can also convert the output SAI files to SAM files. Assuming you are in the directory museomics/part2/6_bwa, run:
# 28S
mkdir 28s; cd 28s
# Align reads
bwa aln -l 1024 -n 0.01 -t 20 ../bwa_index/Dmicrocephalus_28s/Dmicrocephalus_28s ../../5_fastqscreen/Drhea_tally.tagged_filter.fastq > Drhea_28s.sai
# Convert .sai to .sam (bwa samse -f sam index sai fastq)
bwa samse -f Drhea_28s.sam ../bwa_index/Dmicrocephalus_28s/Dmicrocephalus_28s Drhea_28s.sai ../../5_fastqscreen/Drhea_tally.tagged_filter.fastq
# 12S
mkdir ../12s; cd ../12s
# Align reads
bwa aln -l 1024 -n 0.01 -t 20 ../bwa_index/Dmicrocephalus_12s/Dmicrocephalus_12s ../../5_fastqscreen/Drhea_tally.tagged_filter.fastq > Drhea_12s.sai
# Convert .sai to .sam (bwa samse -f sam index sai fastq)
bwa samse -f Drhea_12s.sam ../bwa_index/Dmicrocephalus_12s/Dmicrocephalus_12s Drhea_12s.sai ../../5_fastqscreen/Drhea_tally.tagged_filter.fastq
# RAG1
mkdir ../rag1; cd ../rag1
# Align reads
bwa aln -l 1024 -n 0.01 -t 20 ../bwa_index/Dmicrocephalus_rag1/Dmicrocephalus_rag1 ../../5_fastqscreen/Drhea_tally.tagged_filter.fastq > Drhea_rag1.sai
# Convert .sai to .sam (bwa samse -f sam index sai fastq)
bwa samse -f Drhea_rag1.sam ../bwa_index/Dmicrocephalus_rag1/Dmicrocephalus_rag1 Drhea_rag1.sai ../../5_fastqscreen/Drhea_tally.tagged_filter.fastq
cd ..
Finally, assuming you are in the directory museomics/part2/6_bwa, we will compress SAM files to BAM and remove unmapped reads.
conda activate samtools
cd 28s
# Convert .sam to .bam
samtools view --threads 4 -S -bh Drhea_28s.sam > Drhea_28s_mapANDunmap.bam
# Remove unmapped reads
samtools view --threads 4 -bh -F 4 Drhea_28s_mapANDunmap.bam > Drhea_28s_map.bam
cd ../12s
# Convert .sam to .bam
samtools view --threads 4 -S -bh Drhea_12s.sam > Drhea_12s_mapANDunmap.bam
# Remove unmapped reads
samtools view --threads 4 -bh -F 4 Drhea_12s_mapANDunmap.bam > Drhea_12s_map.bam
cd ../rag1
# Convert .sam to .bam
samtools view --threads 4 -S -bh Drhea_rag1.sam > Drhea_rag1_mapANDunmap.bam
# Remove unmapped reads
samtools view --threads 4 -bh -F 4 Drhea_rag1_mapANDunmap.bam > Drhea_rag1_map.bam
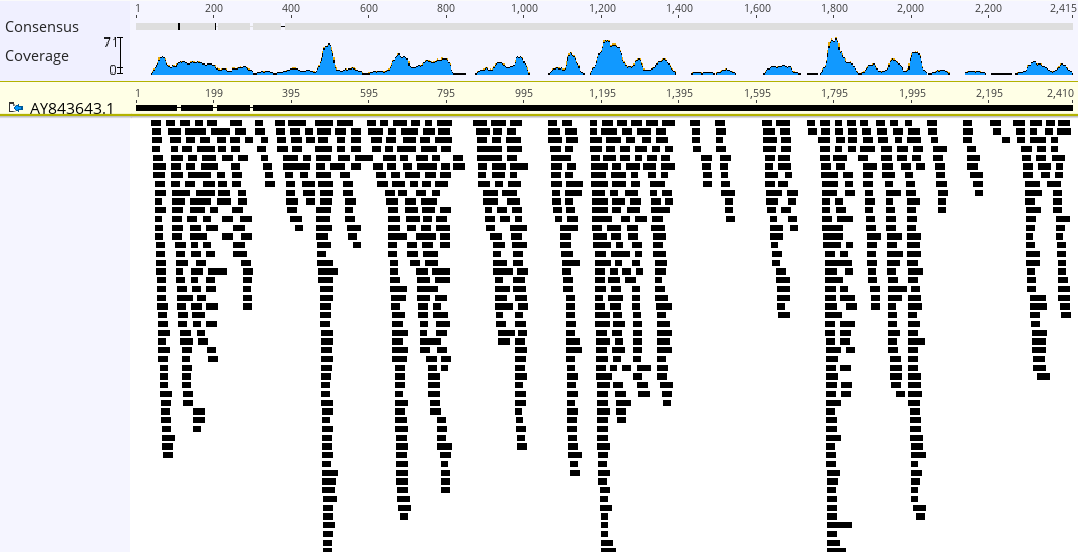
Exercise 5 The command
samtools stats FILE.bam > FILE.txtprovides basic statics about a BAM file (read alignment). Compare the BAM files generated for 12S, 28S, and RAG1. Create a hypothesis to explain the differences in number of mapped reads between nuclear and mitochondrial genes.Show answer
In terms of the number of mapped reads, the observed pattern was 12S > 28S > RAG1. This can be explained primarily by differences in gene copy numbers rather than reference length. Although 28S is physically longer than 12S, the latter exhibited much higher sequencing depth. This is due to the presence of multiple mitochondria per cell, which significantly increases the likelihood of capturing mitochondrial DNA (mtDNA) fragments. Similarly, 28S yielded more reads than the nuclear RAG1 gene because the 28S rRNA gene exists in multiple tandem copies within the nucleolus, whereas RAG1 is typically present as a single-copy gene.
Reference bias
Reference bias is an error where the mapped reads are reference-dependent. In museomics, this is particularly dangerous because hDNA is very short. If a read has too many differences from the reference, the aligner decides it’s “too messy” and throws it away or assigns it a low Mapping Quality (MapQ). As such, the final results will look “more like the reference” than the actual sequenced organism.
Although out of the scope of this short workshop, possible solutions for reference bias include:
- (1) running baiting and iterative mapping (e.g. MITObim), so that reference bias can emerge in the first iteration, but new references are created iteratively based on the contigs of previous iteration.
- (2) running multiple linear mappings against different references, aligning output consensus sequences, and masking ambiguities with IUPAC N;
- (3) running a multiple sequence alignment (MSA) of potential references, calling the consensus of the MSA and coding SNPs with IUPAC ambiguities, and mapping reads against this synthetic reference (suggested in Oliva et al. 2021);
- (4) running multiple linear mappings against different references, selecting best mappings using mapping quality indexes, merging mappings via lift over procedure (developed by Chen et al. 2021)
- (5) building variation graph from different references and mapping reads against the graph (see tutorials here and here).
Post-processing
Consensus calling
# Create a new directory
mkdir ../7_consensus; cd ../7_consensus
conda activate samtools
# BWA: 12S
samtools sort ../6_bwa/12s/Drhea_12s_map.bam | samtools consensus -f FASTA - -o Drhea_12s_consensus.fas
# BWA: 28S
samtools sort ../6_bwa/28s/Drhea_28s_map.bam | samtools consensus -f FASTA - -o Drhea_28s_consensus.fas
# BWA: RAG1
samtools sort ../6_bwa/rag1/Drhea_rag1_map.bam | samtools consensus -f FASTA - -o Drhea_rag1_consensus.fas
Exercise 6 There are strings of contiguous IUPAC N in the consensus sequences. Is IUPAC N the same as missing data? Why is IUPAC N common in museomics?
Show answer
IUPAC N is common in museomics due to low coverage.IUPAC N and question marks ? (missing data) are not the same. IUPAC N represents any nucleotide, whereas a question mark represents any nucleotide or a gap. The user can specify whether low coverage will be coded as N or ?, but the common approach is coding it with N. However, coding it with N is less coservative (more assumptions) than coding it with question marks.
Blast
BLAST (Basic Local Alignment Search Tool) is one of the most fundamental algorithms in bioinformatics. It is used to compare a “query” sequence (DNA, RNA, or protein) against a massive database of known sequences to find regions of similarity. That is, Blast is a quick method to identify sequences.
- Visit https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome
- Copy the consensus sequences generated in this tutorial in “Enter query sequence” and clicl on BLAST.
- Check the identity of each sequence